BandLab Library
Documentation
Table of Content
2. Setting Atomic
Data: Overview
3. Setting Hopping
Integrals: Overview
4. Setting
Correlations: Overview
5. Computing
Physical Properties: Overview
6. Using Materials
Data Base: Overview
A.1 Computing
Density of States
A.2 Computing
Energy Bands and Partial Characters
A.5 Computing
Atoms & Structure
A.6 Computing
Hoppings Integrals
B.1 Visualizing
Density of States
B.2 Visualizing
Energy Bands and Partial Characters
B.3 Visualizing
Fermi Surfaces
B.6 Visualizing
Full Potential
B.7 Visualizing
Charge Density
B.8 Visualizing
Atoms & Structure
C.1 Accessing
Atoms General Properties
C.2 Accessing
Atoms Structural Properties
C.3 Accessing
Atoms Common Tasks
D.1 Setting
General Properties for an Atom..
D.2 Setting
Magnetic Properties for an Atom..
D.3 Setting
Valence State Properties for an Atom
D.4 Setting
Semicore State Properties for an Atom
D.5 Setting Deep
Core State Properties for an Atom
E.1 Setting
General Properties for Tight-Binding State
E.2 Setting Rotate
Properties for Tight-Binding State
E.3 Setting
General Properties for Correlated State
E.4 Setting Rotate
Properties for Correlated State.
E.5 Setting
Correlation Properties for Correlated State
F.2 Accessing
Method Exchange Correlation Functional
F.3 Accessing
Method Convergency Criteria
F.4 Accessing
Method Mixing Criteria
F.6 Accessing K
integration options
F.7 Accessing
Plane wave FFT options
© Copyright 2005
1. Ove rview
of BandLab
Welcome to BandLab, the scientific software for Windows systems that
performs electronic structure calculations of crystalline solids. BandLab is a
library dynamically linked to MStudio. BandLab also uses MScene library for
visualization.
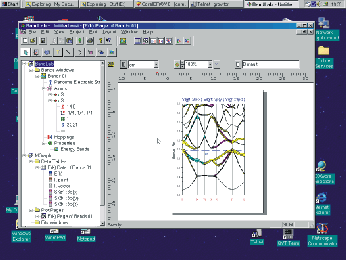
BandLab consists of a window that is used to set up input data, perform
calculations and analyze output data. The BandLab Window, shortly Bands Window
is called using Bands command of the Project menu or corresponding
button of the Bands Toolbar. The Bands
window consists of several Property Pages:
- Atoms Page
- Hoppins Page
- Hubbards Page
- Properties Page
A lower area of the Bands Window is an output area where the output of
the calculation is shown.
 The engine of the BandLab is a program called LmtART. The LmtART is
an executable file that reads the input data, performs band structure
calculation, and stores the output files. The Bands Window only controls this
process, it prepares input for the LmtART using dialog windows, and starts
LmtART program as a separate thread. When the calculation is finished, LmtART
notifies the BandLab, and the output files are read by the Bands Window and can
be visualized by simple mouse click operations.
The engine of the BandLab is a program called LmtART. The LmtART is
an executable file that reads the input data, performs band structure
calculation, and stores the output files. The Bands Window only controls this
process, it prepares input for the LmtART using dialog windows, and starts
LmtART program as a separate thread. When the calculation is finished, LmtART
notifies the BandLab, and the output files are read by the Bands Window and can
be visualized by simple mouse click operations.
While running LmtART program for complicated compounds can be rather
slow (it depends how many atoms per unit cell is chosen), the BandLab can be
used to prepare the input files, after which they can be copied to another
computer where the LmtART can be run in a faster way. After run is performed,
the output files can be copied back to the PC and BandLab can analyze them
quickly. All input/output files are formatted, and therefore system
independent.
The BandLab uses scratch directory to run LmtART program. By default
scratch directory is called LmtRUN. It exists at the MStudio installation
directory. All input/output files prepared by the Bands window are stored in
this directory.
A separate issue of the BandLab is its Materials Database. The BandLab
has a database of compounds that have been calculated using the LmtART program.
The database is an important part of the library, it can be called by pressing DataBase
button located on the top right part of the Bands Window. The input/output
files can be stored within the database: this gives possibility to use these
files on the remote machines. Refer to Using DataBase topic for
the detailed discussion.
2. Setting Atomic Data: Ove rview
Atomic data set-up is the primary set of data describing the crystalline solid. The first property page of the Bands Window is designed to describe these parameters:
- Lattice parameter in atomic units
- B/A and C/A ratios if the lattice has
orthorhombic distortions
- Primitive translations in units of lattice
parameter
- Spin polarization, if necessary
- Spin-orbit coupling, if necessary
- Temperature.
More options can be
accessed via clicking Access All Options button. The corresponding
property pages are
- General Properties
- Structure Properties
- Common Tasks Properties
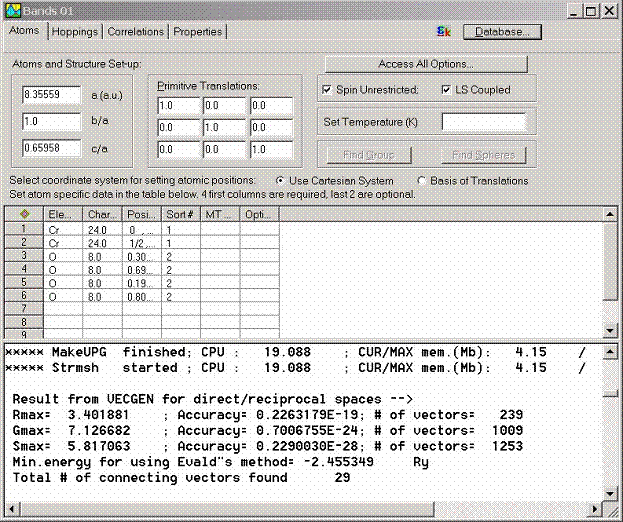
The following set of
data has to be given for each atom in the unit cell
- Element title, like Na, Nb, etc.
- Nuclear charge. This is the most important
parameter over which the software recognizes the atoms.
- Position of atom in the unit cell in the
units of lattice parameter.
- If several atoms have the same type,
specify it in the Sort column. This will be used to determine
crystal group. For example, the compound YBa2Cu3O7 has 12 atoms. At the
same time there only 8 nonequivalent atoms: Y, Ba, Cu1, 2*Cu2, 2*O1, 2*O2,
2*O3, and O4. Therefore, sort column will be 1 for Y@1, 2 for Ba@2, 2 for
Ba@3, 3 for Cu1@4, 4 for Cu2@5, 5 for O1@6, 5 for O1@7, 6 for O2@8, 6 for
O2@9, 7 for O3@10, 7 for O3@11, 8 for O4@12. Here El@N shortly denotes the
element at position N.
- The radius of the MT sphere can be
optionally specified if this value is known. If this value is unknown, it
can be computed by the LmtART.
Additional options for atoms can be accessed by highlighting the entire
row and calling the Properties dialog box with the right mouse button.
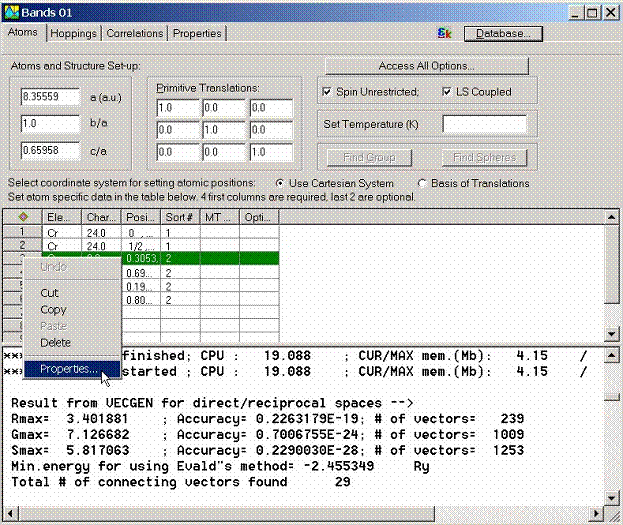
Another possibility to call the Atom Properties dialog box is to access
it from the Object Explorer.
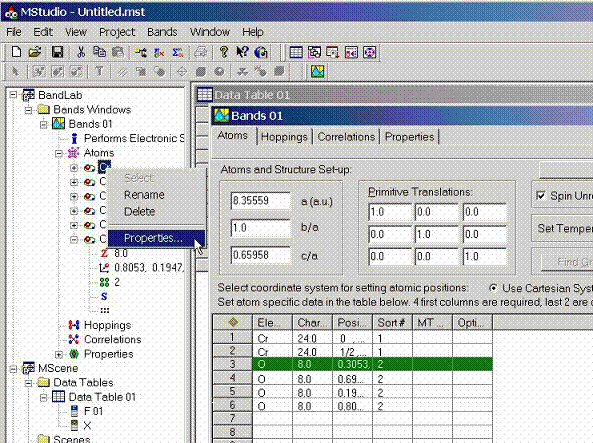
The dialog box which allows to access various properties for the selected atom appears. These properties include:
- General Properties
- Magnetic properties
- Valence States proeprties
- Semicore States properties
- Deep core states properties
3. Setting Hopping Integrals: Ove rview
Hopping integrals are necessary if tight-binding (possibly non-orthogonal) calculations of the electronic structure are desired. Tight-binding picture is an important step in understanding chemical bonding of the materials. After performing full band structure calculations, construction of simple tight-binding models describing complicated spaghetti of bands is extremely useful.

The BandLab offers two
ways of working with tight-binding.
- The first way is to perform band structure
calculation and withdraw tight-binding integrals. This can be performed
using so called tight-binding LMTO representation used by the LmtART
program. A drawback of this approach is that too many hopping integrals
are usually withdrawn; therefore manual analysis is required after that to
select only important hoppings. Advantage of this approach is that the
correct orders of magnitudes for all hoppings are obtained.
- The second way is to enter hopping
integrals from the terminal and try to fit the electronic structure by
changing the input values Advantage of this approach is that only several
hoppings integrals are varied. A drawback of it is that the significant
hoppings have to be known before the analysis is performed.
Both ways require
quick recalculation and drawing of the band structure using a set of hoppings,
and the BandLab offers the fastest way of operating with these problems.
Usually, if building
tight-binding picture for the material is necessary, one first performs the
computation of all possible hopping integrals, and then manually selects the
largest hoppings. The unimportant hoppings can be erased from the hoppings
table and few-parameter model can be constructed.
The following inputs
are required to work with the hoppings:
- Cluster size: this is the size of the
nearest neighbor sphere surrounding every atom. Normally only first
nearest neighbors are important to consider. The hoppings between these
nearest neighbors will be considered.
- Filling: this is number of electrons to
find the position of the Fermi level.
- Tight-binding basis: set orbitals which
represent tight-binding basis. For example, for CuO2 plane of YBa2Cu3O7,
3d electrons of Cu and 2p electrons of O should be considered. Syntax used
for setting the orbital is El@N::nl with El is an element title, N is the
position number, n is the main quantum number and l is the orbital quantum
number. For example CuO2 plane is represented by Cu@1::3d orbital and
O@2::2p, O@3::2p oxygen orbitals.
- The use of the local coordinate system for
every atom can be set in the following columns of this table. Refer to the
LmtART manual for the detailed set-up of these data.
- Irreducible hopping integrals have to be
specified in the lower table. Term irreducible refers to those hoppings
that cannot be obtained by applying group operation. In order to find
irreducible hoppings automatically for given cluster and orbitals, click Irreducible
button. Note that the cluster size and the orbitals have to be set up
while the hopping table should be completely empty in order to perform
this operation. (You should manually erase all hoppings from the table if
its contents is not empty since the program does not overwrite the
existing data.)
- The hoppings are given FROM a particular
orbital TO another orbital VIA connection vector. The value of hopping and
possibly the overlap integral can be specified. Term orbital here refers
to a particular lm-orbital. These,
for example, will be Cu@1::3d{xy}, Cu@1::3d{yz}, Cu@1::3d{zx},
Cu@1::3d{x2-y2}, Cu@1::3d{3z2-1}, total 5 orbitals for Cu 3d basis. If the connection vector is set to 0,0,0
the hopping is reduced to the level position for this particular orbital.
The overlap integral can be set to 1 in this case. For non-zero
connections, the overlap integral can be set to zero if orthogonal
tight-binding is considered.
It is important to understand that the tight-binding basis in this set-up considers all (2l+1) orbitals for given l-shell, not only selected components. This is done for easy transformation between cubic and spherical harmonics as well as to perform rotations to local coordinate systems. In the example of CuO2, it is well known that only Cu@1::3d{x2-y2}orbital is in fact the only important one. To take this into account shift the positions of other orbitals, Cu@1::3d{xy}’s and Cu@1::3d{3z2-1} by some amount either above or below, and do not specify hoppings from that orbitals. They will be resolved as levels and will not play any role in the tight-binding model constructed.
For spin-polarized and possibly spin-orbit coupled set-ups, the
following syntax for orbitals definitions should be used: El@N-up::nl{…} or
El@N-dn::nl{…}. These are, for example, Cu@1-up{x2-y2} and Cu@1-dn{x2-y2}
orbitals.
Note finally that if the hopping integral is not specified within the
table, its value is assumed to be equal zero.
The properties for the tight binding orbitals can be accessed via calling
Properties Dialog Box. To call this dialog box select given tight-binding state
and call pop-up menu by the right mouse button.
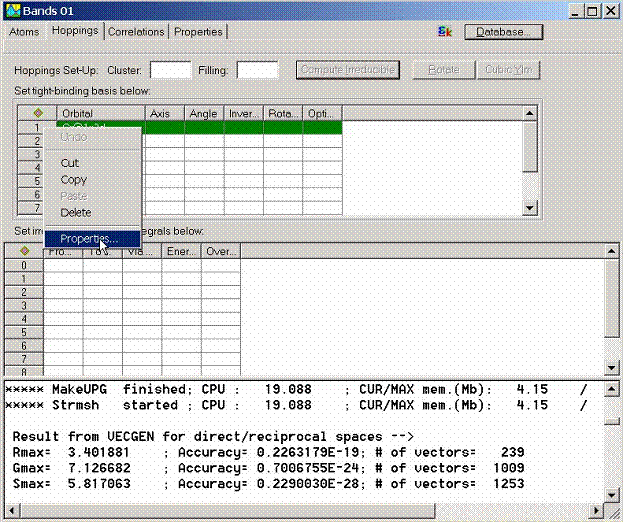
Another way to call
this dialog box is to find the tight binding state at the object explorer and
double click on it or call the pop-up menu using the right mouse button.

The dialog box which
allows to further customize the properties of the tight-binding state will
appear. It contains the following property pages
- General Properties
- Rotate Properties
4. Setting Correlations: Ove rview
Correlations among d- or f-electrons in solids are important for determining their physical properties. Local density and generalized gradient approximations do not treat properly systems with the correlated electrons. A classical example of the failure is Mot-Hubbard insulator NiO. BandLab currently offers LDA+U scheme for including the effects of strong correlations into account. way of operating with these problems.
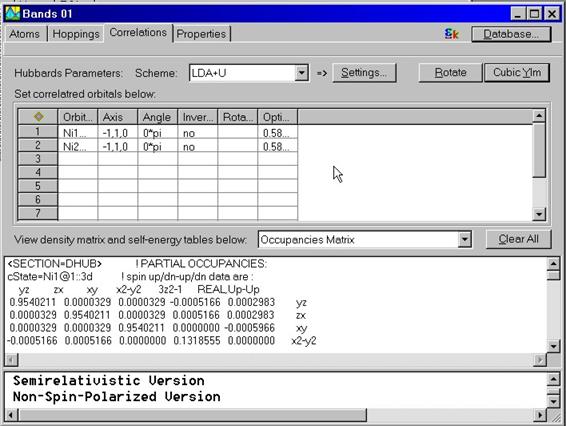
The following inputs
are required to set up correlations:
- Basis set for correlated orbitals: set
orbitals which represent tight-binding basis. For example, for
Mott-Hubbard insulator NiO, 3d
electrons of Ni can be considered. Syntax used for setting the orbital is
the same as for setting hopping integrals.
El@N::nl with El is an element title, N is the position number, n
is the main quantum number and l is the orbital quantum number. For
example Ni 3d orbitals are represented by Ni@1::3d and Ni@2::3d.
- The use of the local coordinate system for
every atom can be set in the following columns of this table. Refer to the
LmtART manual for the detailed set-up of these data.
- Slater integrals should be specified in
the Options column. There must be total l+1 number for given l-orbital.
For d-electrons these are F0, F2, and F4 parameters.
The properties for the correlated orbitals can be accessed via calling
Properties Dialog Box. To call this dialog box select given correlated state and
call pop-up menu by the right mouse button.
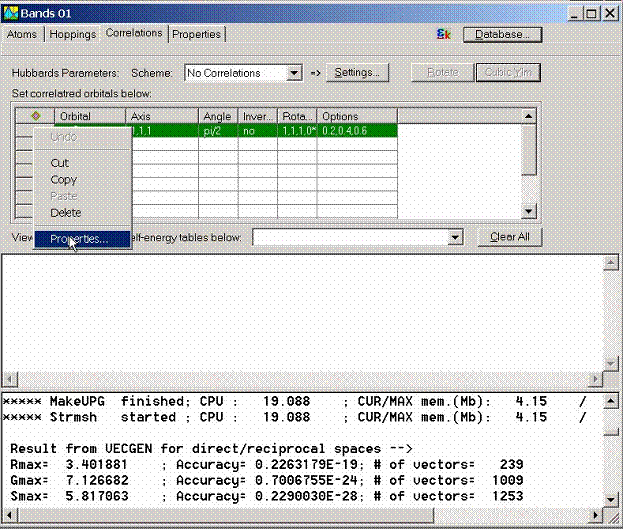
Another way to call
this dialog box is to find the tight binding state at the object explorer and
double click on it or call the pop-up menu using the right mouse button.

The dialog box which
allows to further customize the properties of the correlated state will appear.
It contains the following property pages
- General Properties
- Rotate Properties
- Correlations Properties
When self-consistent calculation using LDA+U is done, a set of density matrices and orbital-dependent corrections to the potential are calculated. They can be analyzed in the lower part of the Correlations window.
5. Computing Physical Properties: Ove rview
Computation of physical properties is the goal of the BandLab library.
The LmtART
program is the executable module that does this calculation. It reads input
files prepared by the bands Window, performs the calculation of the desired
property, and notifies the Bands Window again that the calculation is finished.
The BandLab reads the output files and visualizes them with help of the MScene
library.
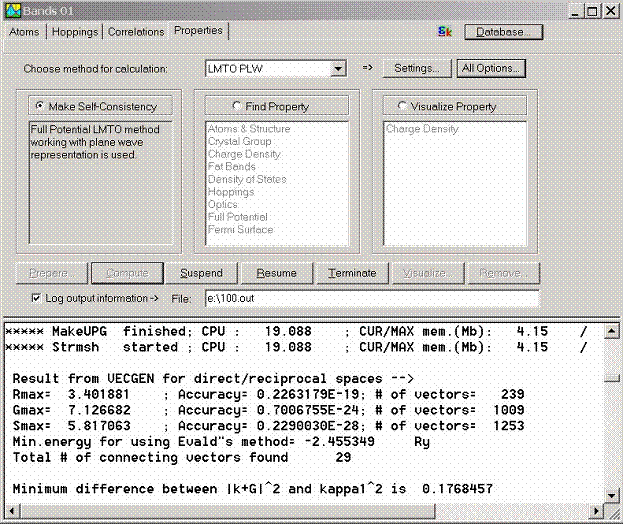
The purpose of the Properties page of the Bands Window is to make final
preparations before starting LmtART, start LmtART code, and visualize the
output data computed by the LmtART. Once the compound data, and, optionally,
hopping integrals are specified using Atoms, Hoppings and Correlations property
pages, the last step is to choose the method of computing the physical
property.
Three methods are
currently available within the use of BandLab library:
- Full potential LMTO method for electronic
structure calculation. This is the most precise approach for computing
physical properties such as one-electron structure, but at the same time,
it is computationally slow approach.
- ASA-LMTO method for electronic structure
calculation. This is a fast method, but less accurate comparing to the
full potential LMTO method.
- Tight-binding method. This method uses the
hopping integrals given in the Hoppings property page and can be
used to solve few-band model Hamiltonians.
Each method has its own set of options which can be set by clicking Method
Settings A more complete set of options can be accessed via clicking All
Options button. It includes
- Control
parameters
- Exchange
correlation choice
- Convergence
criteria
- Mixing
options
- LMTO options
- K-intergration
settings
- Plane wave
fast fourier transform settings
Both full potential LMTO and ASA LMTO methods require self-consistent
calculation to be performed before computing any physical property such as the
band structure or density of states. To make self-consistency, switch on radio
button Make Self-Consistency and click Compute button. The
self-consistent calculation will start. The output window in the lower part of
the Bands Window will show haw the self-consistent process is performed. This
however maybe rather slow process especially when compounds with many atoms are
considered. At the end of the calculation, the framework creates the file
consisting the self-consisting charge density.
Once self-consistency is performed, such physical properties as Fat
Bands analysis or Density of States can be computed. One can also compute
Hopping integrals necessary to build the tight-binding model.
To compute physical property such as Fat Bands, set radio button Find
Property to the On position.
Chose Fat Bands property from the drop-down list and press Compute
button. ( You may also use Prepare button to set more options before
pressing compute button.)
The following set of
properties can be computed by the BandLab:
- Atoms & Structure
- Crystal Group
- Charge Density
- Fat Bands
- Density of States
- HoppingsHIDD_COMPUTE_HOP
- OpticsHIDD_COMPUTE_OPT
- Full Potential
- Fermi Surface
After the computation of a particular property is finished, the
framework will add this into the computed properties list of the Visualize
Property drop-down list.
To start visualizing the properties that have been computed, choose
radio button Visualize Property. Highlight corresponding property and
press Visualize button. A visualization dialog box appears. You can
start visualizing the data by pressing Visualize with the current settings
button in the lower part of this dialog box.
Such features as band characters can be visualized readily by the
BandLab. The visualization dialog box contains a tree of atomic orbitals whose
characters can be visualized within the band structure plot. To be able to
visualize a particular character, drug the corresponding icon and drop it into
the tree item describing the plot such as E(k) or DOS. You can choose several
orbitals to be visualized at the same time. After, the band characters to be
visualized are selected, press Visualize with the current settings
button in the lower part of this dialog box. The Plot Page with the details of
the band structure and the characters should appear.
The following controls
help to visualize the computed properties:
- Visualzing Atoms & Structure
- Visualizing Crystal Group
- Visualizing Density of States
- Visualizing Energy Bands and Partial
Characters
- Visualizing Fermi Surfaces
- Visualizing Optics
- Visualizing Full Potential
- Visualizing Charge Density
All visualization
plots can be additionally customized using the commands of MScene library.
Refer to the MScene Library documentation for corresponding instructions.
6. Using Materials Data Base: Ove rview
Materials database is important part of the BandLab library. Instead of maintaining its own input files, one can use database to extract set-ups for similar materials and then slightly modify them. After computation of the material properties and or self-consistency process is performed one can store the computed data into the database to be able to use them in the future runs.
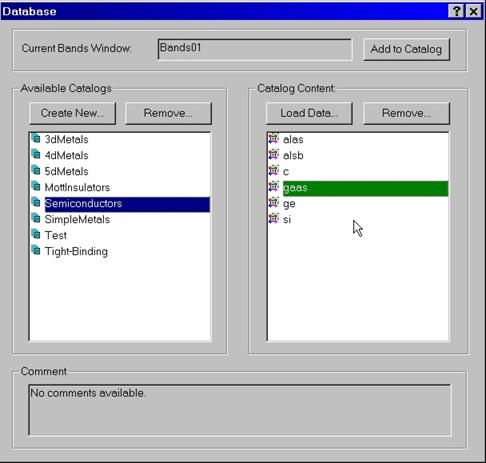
To call database press Database button located at the top right
part of the bands window. The database dialog box appears. The database
consists of catalogs and their contents. A set of predefined catalogs
describing set-ups for 3d, 4d, 5d, simple metals, etc is provided with this
installation.
To load data from the database into the Bands Window select a particular
compound and press Load Data button. To store data computed by the Bands
Window, use button Add to Catalog located on the top of the database
dialog box.
New catalogs can be created and removed. Entries within the catalog can
also be removed using corresponding buttons.
In fact, the database maintained by the framework is located in the
database folder of the directory where installation of MStudio has been
performed. Navigate database folder, the input and output files for many
different examples are stored in its subdirectories. All these files are either
input or output files of the LmtART program that performs actual computations.
Refer to the LmtART manual for detailed instructions about input and output to
this program.
7. LmtART program
LmtART is free scientific software designed to perform electronic structure calculations of the materials. It is written using Fortran 90 programming language and uses full dynamical memory model. The source codes for that program, as well as installation and operating instructions can be downloaded from the same site as the MStudio, MScene and BandLab program. Refer to specific license agreement and copyright notice when using LmtART codes.
A.1 Computing Density of States
To compute density of states
you need to enter minimum and maximum energies in either Ry or eV as well as #
of intervals to divide the energy range Emax – Emin. You also have an option to
enter # of divisions for the Brillouin zone, e.g., 10,10,10 which supersedes
the input given an
The program computes and stores the file with an extension .dos which can be further visualized.
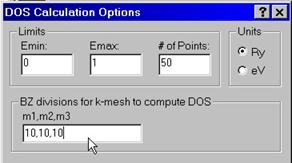
A.2 Computing Energy Bands and Partial Characters
Energy bands can be computed along high-symmetry directions. For simple lattices like bcc,fcc, hcp, simple cubic, etc. these high symmetry directions are listed in the files str.* located in the atomdat directory. To compute energy bands and partial characters you need to enter # the number if intervals to divide every high-symmetry line. The default # is 20. If you wish to compute the energy bands in a set of custom directions, use the corresponding table control to enter Cartesian coordinates for every k-point and their notations. For example, enter in each row: g 0,0,0 ; X 0,0,1, L 0.5,0.5,0.5 to compute the energy bands along g-X-L direction. Tip: use g to denote G point. When entering the coordinates, do not take into account b/a, c/a ratios, they will be accounted for automatically.
The program computes and stores the file with an extension .fat which can be further visualized.

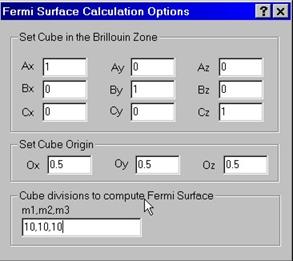
A.3 Computing Fermi Surfaces
To compute Fermi surface you need to set up a cube in the reciprocal space for which the grid of k-points will be generated and the band structure will be computed as Ej(kx,ky,kz). Then, the software will draw a cross-section Ej(kx,ky,kz)=Ef for all bands j which cross the Fermi level.
To set up the cube, enter three vectors defining the cube (e.g., 100,010,001) and the origin point (e.g. 0.5,0.5,0.5). Every k-point will be computed as the cube point minus the origin vector. Note that b/a and c/a ratios will be taken into account automatically.
You also have an option to
enter # of divisions for the Brillouin zone, e.g., 10,10,10 which supersedes
the input given an
The program computes and stores the file with an extension .frs which can be further visualized.
A.4 Computing Crystal Group
To compute crystal group, there is no optional input parameters. The program stores file with the extension .grp which can be further visualized.
A.5 Computing Atoms & Structure
To compute atoms & structure, there is no optional input parameters. The program stores file with the extension .atm which can be further visualized.
A.6 Computing Hoppings Integrals
To compute atoms & structure, there is no optional input parameters. The program computes hoppings table and overwrites the table given within the bands window.
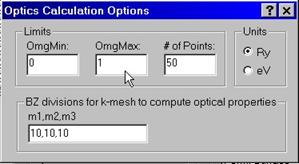
A.7 Computing Optics
To compute optical frequencies
you need to enter minimum and maximum frequencies in either Ry or eV as well as
# of intervals to divide the energy range Emax – Emin. You also have an option
to enter # of divisions for the Brillouin zone, e.g., 10,10,10 which supersedes
the input given an
The program computes and stores the file with an extension .opt which can be further visualized.
A.8 Computing Full Potential
To compute full potential, there is no optional input parameters. The program stores file with the extension .pot which can be further visualized.
A.9 Computing Charge Density
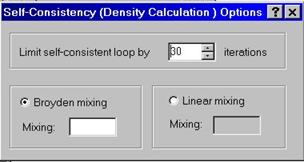
Charge density is computed self-consistently, therefore this is the most time-consuming step in the calculation. You need to enter total # of iterations to perform the self-consistent calculation as well as mixing scheme. You have an option to select Broyden mixing which usually speeds up the self-consistent cycle significantly. If you feel this scheme does not work and you cannot get convergent result, use linear mixing scheme.
B.1 Visualizing Density of States
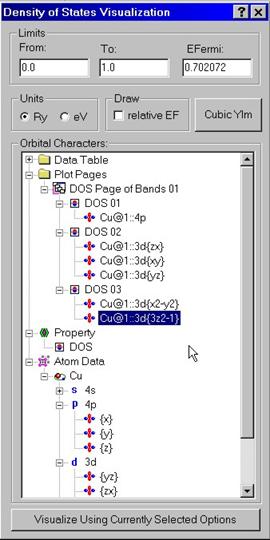
In order to visualize density of states and partial contributions, use Visualize Density of States dialog box. Enter lower and upper energy limits as well as the position of the Fermi energy in either Ry or eV. A special tree control exists to set up which partial characters should be drawn on top of the density of states. Drag partial characters from the Atomic Data folder and drop them within the Page folder into the icon showing the DOS property. Note that you can use right mouse button to access to the pop-up menu with Rename, Delete, Add commands. You can add new Pages and drag calculated properties to every of these pages. You can also put partial characters into the calculated properties by dragging them from the Atomic Data folder. In this way, you can set up spontaneously several pages with each page containing several graphs. When you are done with the descriptions of graphs, press Visualize button at the button. The program will use the graphical library to draw selected set of properties.
B.2 Visualizing Energy Bands and Partial Characters
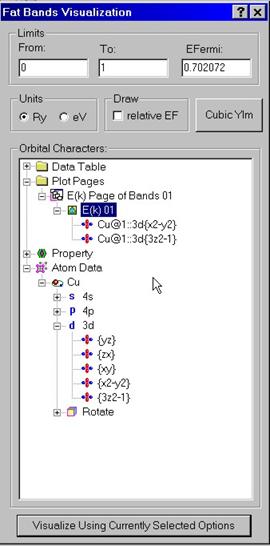 In
order to visualize energy bands and their partial characters, use Visualize
Energy Bands dialog box. Enter lower and upper energy limits as well as the
position of the Fermi energy in either Ry or eV. A special tree control exists
to set up which partial characters should be drawn on top of the energy bands.
They will be shown as fat lines. Drag partial characters from the Atomic Data
folder and drop them within the Page folder into the icon showing the E(k)
property. Note that you can use right mouse button to access to the pop-up menu
with Rename, Delete, Add commands. You can add new Pages and drag calculated
properties to every of these pages. You can also put partial characters into
the calculated properties by dragging them from the Atomic Data folder. In this
way, you can set up spontaneously several pages with each page containing
several graphs. When you are done with the descriptions of graphs, press Visualize
button at the button. The program will use the graphical library to draw
selected set of properties.
In
order to visualize energy bands and their partial characters, use Visualize
Energy Bands dialog box. Enter lower and upper energy limits as well as the
position of the Fermi energy in either Ry or eV. A special tree control exists
to set up which partial characters should be drawn on top of the energy bands.
They will be shown as fat lines. Drag partial characters from the Atomic Data
folder and drop them within the Page folder into the icon showing the E(k)
property. Note that you can use right mouse button to access to the pop-up menu
with Rename, Delete, Add commands. You can add new Pages and drag calculated
properties to every of these pages. You can also put partial characters into
the calculated properties by dragging them from the Atomic Data folder. In this
way, you can set up spontaneously several pages with each page containing
several graphs. When you are done with the descriptions of graphs, press Visualize
button at the button. The program will use the graphical library to draw
selected set of properties.
B.3 Visualizing Fermi Surfaces
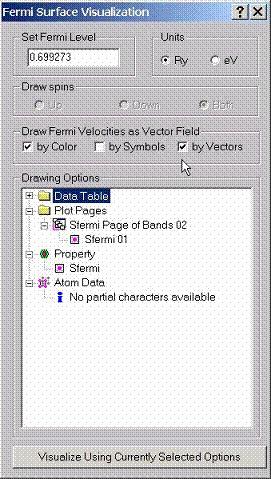 In
order to visualize Fermi surface, use Visualize Fermi Surface dialog
box. Enter the position of the Fermi energy in either Ry or eV. Fermi velocities can be shown either by color
on top of the surface, or by vectors. When you are done with the description of
graphs, press Visualize button at the button. The program will draw a
multi-sheet cross-section showing the solution of the equation Ej(kx,ky,kz)=Ef.
In
order to visualize Fermi surface, use Visualize Fermi Surface dialog
box. Enter the position of the Fermi energy in either Ry or eV. Fermi velocities can be shown either by color
on top of the surface, or by vectors. When you are done with the description of
graphs, press Visualize button at the button. The program will draw a
multi-sheet cross-section showing the solution of the equation Ej(kx,ky,kz)=Ef.
B.4 Visualizing Crystal Group
 The
program shows the table containing the description of the group elements. The
description for every element contains a rotation by an angle around an axis as
well as whether inversion is applied after the rotation. For non-symmorphic
groups a shift may also exist which is applied after the rotational operation.
To see schematically the group element press the row button within the group
table.
The
program shows the table containing the description of the group elements. The
description for every element contains a rotation by an angle around an axis as
well as whether inversion is applied after the rotation. For non-symmorphic
groups a shift may also exist which is applied after the rotational operation.
To see schematically the group element press the row button within the group
table.
B.5 Visualizing Optics
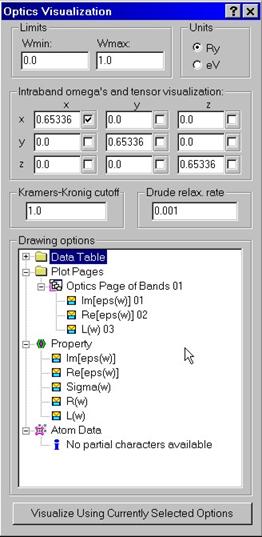 In
order to visualize optical properties, use Visualize Optics dialog box.
Set lower and upper frequency limits for optics visualization in either Ry or
eV. Since intraband contribution involves calculated plasma frequencies, you
can alter them using the corresponding edit fields. Dielectric function is in
general a matrix with the Cartesian components xx,xy,xz,yx,yy,yz, zx,zy,zz.
Check corresponding boxes to set which of the components should be visualized.
To compute real part of the dielectric function, Kramers-Kronig relationship is
used. You have an option to enter upper frequency limit to use in the
Kramrers-Kronig relationship. You have also an option to use a finite
relaxation rate to be used in the Drude intraband contribution. When you are
done with the description of graphs, press Visualize button at the
button.
In
order to visualize optical properties, use Visualize Optics dialog box.
Set lower and upper frequency limits for optics visualization in either Ry or
eV. Since intraband contribution involves calculated plasma frequencies, you
can alter them using the corresponding edit fields. Dielectric function is in
general a matrix with the Cartesian components xx,xy,xz,yx,yy,yz, zx,zy,zz.
Check corresponding boxes to set which of the components should be visualized.
To compute real part of the dielectric function, Kramers-Kronig relationship is
used. You have an option to enter upper frequency limit to use in the
Kramrers-Kronig relationship. You have also an option to use a finite
relaxation rate to be used in the Drude intraband contribution. When you are
done with the description of graphs, press Visualize button at the
button.
B.6 Visualizing Full Potential
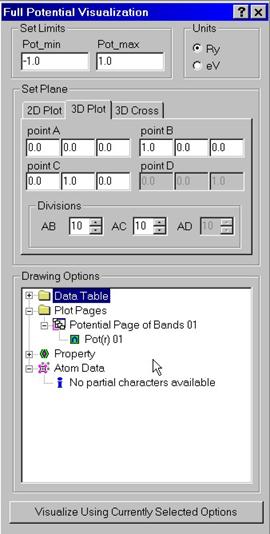 Full
potential can be visualized either along a set of lines, within a plane as a 2D
surface or as a cross-section which is the solution of the equation V(r)=const.
For spin polarized calculations you can also draw exchange-correlation magnetic
fields. Without spin-orbit coupling there is only field along z-direction which
is the difference between spin up and spin down potentials. If spin-orbit
coupling is taken into account there are also fields along x, and y directions.
Full
potential can be visualized either along a set of lines, within a plane as a 2D
surface or as a cross-section which is the solution of the equation V(r)=const.
For spin polarized calculations you can also draw exchange-correlation magnetic
fields. Without spin-orbit coupling there is only field along z-direction which
is the difference between spin up and spin down potentials. If spin-orbit
coupling is taken into account there are also fields along x, and y directions.
To set up a set of lines, plane or a cube, use Cartesian coordinates for the points A, B, C and D. Enter # of divisions to divide intervals AB, AC, (for the line and the plane) and AD (for the cube). Note that b/a and c/a ratios will be taken into account automatically.
The program will draw a 2D surface V(x,y). for the PLANE option. Note that we define x-axis here as left right axis, and we define y-axis here as far-near axis. The latter redefines the set of the MScene library where y-axis is defined as bottom –top axis. We use corresponding layer-mapping control to set up proper definitions. The same is true when V(x,y,z)=const. (CUBE option) is used. The program will draw the cross-section within the selected cube. The program also shows the positions of atoms by colored balls.
If magnetic field is calculated, it can be visualized as well. The magnetic field can be shown by color, symbols, or as a vector field on top of the surface. In the latter case, the maximum value of the vector length is calculated for the entire set of data. For a particular data points selected by the surface sometimes it may be too small.
If vectors did not appear, one has to rescale the lengths
of the vectors to achieve a desired effect.
Find the surface at the data explorer and use vectors property page to
rescale their length and customize the appearance.
B.7 Visualizing Charge Density
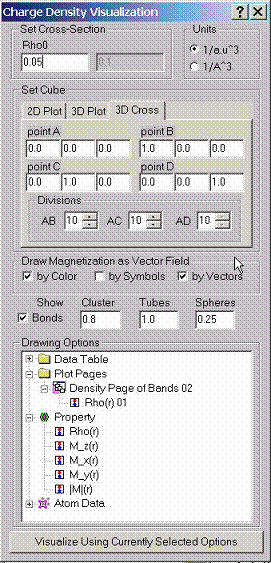 Charge
density can be visualized either along a set of lines, within a plane as a 2D
surface or as a cross-section which is the solution of the equation r(r)=const. For spin polarized calculations
you can also draw magnetization fields. Without spin-orbit coupling there is
only magnetization along z-direction which is the difference between spin up
and spin down densities. If spin-orbit coupling is taken into account there are
also magnetizations along x, and y directions.
Charge
density can be visualized either along a set of lines, within a plane as a 2D
surface or as a cross-section which is the solution of the equation r(r)=const. For spin polarized calculations
you can also draw magnetization fields. Without spin-orbit coupling there is
only magnetization along z-direction which is the difference between spin up
and spin down densities. If spin-orbit coupling is taken into account there are
also magnetizations along x, and y directions.
To set up a set of lines, plane or a cube, use Cartesian coordinates for the points A, B, C and D. Enter # of divisions to divide intervals AB, AC, (for the line and the plane) and AD (for the cube). Note that b/a and c/a ratios will be taken into account automatically.
The program will draw a 2D surface r(x,y). for the PLANE option. Note that we define x-axis here as left right axis, and we define y-axis here as far-near axis. The latter redefines the set of the MScene library where y-axis is defined as bottom –top axis. We use corresponding layer-mapping control to set up proper definitions. The same is true when r(x,y,z)=const. (CUBE option) is used. The program will draw the cross-section within the selected cube. The program also shows the positions of atoms by colored balls.
If magnetization is calculated, it can be visualized as well. The magnetization can be shown by color, symbols, or as a vector field on top of the surface. In the latter case, the maximum value of the vector length is calculated for the entire set of data. For a particular data points selected by the surface sometimes it may be too small.
If vectors did not appear one has to rescale the lengths
of the vectors, to achieve a desired
effect. Find the surface at the data explorer and use vectors property page to
rescale their length and customize the appearance.
B.8 Visualizing Atoms & Structure
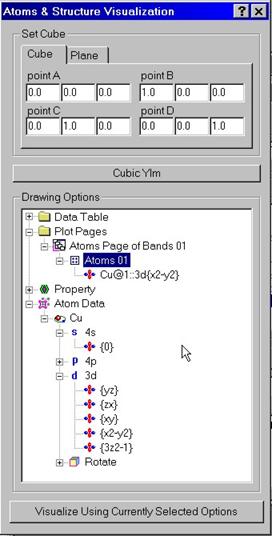 Positions
of atoms and various atomic orbitals can be drawn for a given crystal
structure. The structure can be shown either within a cube or for a plane.
Positions of atoms are shown by colored balls. Every atom of different kind is
colored differently by default. The size of every ball is computed according to
its atomic sphere. If the latter ones are unknown, the balls will be drawn of
the same size.
Positions
of atoms and various atomic orbitals can be drawn for a given crystal
structure. The structure can be shown either within a cube or for a plane.
Positions of atoms are shown by colored balls. Every atom of different kind is
colored differently by default. The size of every ball is computed according to
its atomic sphere. If the latter ones are unknown, the balls will be drawn of
the same size.
Atomic orbitals can be shown at every site of the lattice.
Drag various orbitals from the Atomic Data folder and drop them within the Page
folder into the icon showing the Atoms property. Note that you can use right
mouse button to access to the pop-up menu with Rename, Delete, Add commands.
When visualizing the orbitals atomic balls will be drawn with semitransparency.
C.1 Accessing General Properties
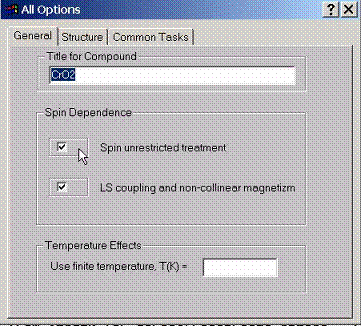 Describe
general properties such as compound title, spin dependence, and whether to
switch relativistic spin orbit coupling effects (available only when spin
unrestricted treatment is used).
Describe
general properties such as compound title, spin dependence, and whether to
switch relativistic spin orbit coupling effects (available only when spin
unrestricted treatment is used).
Also temperature in K can be set which will use finite temperature Fermi Dirac functions in the calculation.
C.2 Accessing Structural Properties
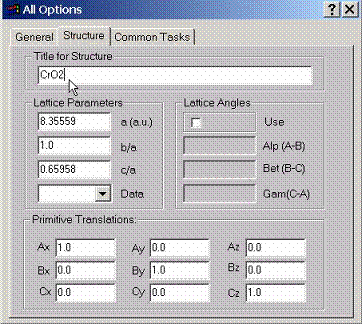 Structural parameters have
to be described using this property page. Lattice parameter should be given in
atomic units. b/a and c/a ratios set scaling of y and z components for
primitive translations. Data box can be used to select primitive translations
for most common structures.
Structural parameters have
to be described using this property page. Lattice parameter should be given in
atomic units. b/a and c/a ratios set scaling of y and z components for
primitive translations. Data box can be used to select primitive translations
for most common structures.
If primitive translations are given by the angles between vectors AB, BC and CA, the translations should only set the lengths for the vectors A, B, C. After the translations read, they first will be translated into cartesian coordinates and then b/a, c/a will be applied along y and z axes, respectively.
C.3 Accessing Atoms Common Tasks
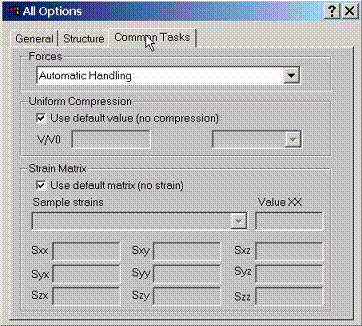 Task
1. The calculation of forces on atoms can be done by the program. Since it is
relatively time consuming calculation due to incomplete basis set corrections,
several options can be choosen: no force calculation (default), fast force
calculation with shorter output, full force calculation with long output of all
partial contributions.
Task
1. The calculation of forces on atoms can be done by the program. Since it is
relatively time consuming calculation due to incomplete basis set corrections,
several options can be choosen: no force calculation (default), fast force
calculation with shorter output, full force calculation with long output of all
partial contributions.
Task 2. If volume compression of the lattice is required, change only the parameter V/V0. Do not change lattice constants, sphere radii and any other data.
Task 3. If straining the lattice is required, specify strain matrix here. Sample strains such as volume conserved stretching are provided. if strain is applied, it is always good idea to first find minimal non-overlapping sphere radii corresponding to the largest strain. Fix the radii and then explore strained lattice for different strains with the fixed spheres.
D.1 Setting General Properties for an Atom
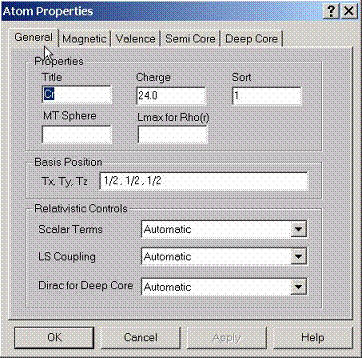 For
each atom, several properties can be specified. The main property is atomic
charge and its position in the lattice. The position can be given in Cartesian
coordinates (Tx, Ty, Tz) or within the coordinate system set by the primitive
translations A, B, C:
For
each atom, several properties can be specified. The main property is atomic
charge and its position in the lattice. The position can be given in Cartesian
coordinates (Tx, Ty, Tz) or within the coordinate system set by the primitive
translations A, B, C:
T=a*A+b*B+c*C.
Sort # should be specified such as different atoms can be of the same kind. This information will be used to find the crystal group.
Scalar relativistic effects can be utilized along with the spin orbit coupling for the valence and semicore states. The deep core states can be found using the full solution of the Dirac equation.
D.2 Setting Magnetic Properties for an Atom
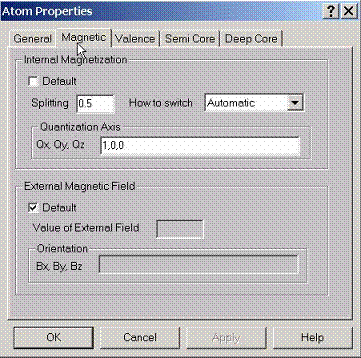 To
find the magnetic spin ordered solutions, the quantization axis and initial
magnetic field can be given. Without spin orbit coupling effects, the
quantization axis is always assumed along z direction. If spin orbit coupling
is switched on, the quantization axis can align along any direction. The intial
magnetic field will be switched on along magnetization axis to kick the atom
out of the paramagnetic state. After that it will be switched off. This initial
magnetic field is simply given by the splitting of the potential along
quantization axis, for z-axis, this is simply the intial difference between
spin up and spin down potentials. Note that if the atom already exists in the
magnetic state, this initial splitting will be set to zero automatically unless
this option is suppressed.
To
find the magnetic spin ordered solutions, the quantization axis and initial
magnetic field can be given. Without spin orbit coupling effects, the
quantization axis is always assumed along z direction. If spin orbit coupling
is switched on, the quantization axis can align along any direction. The intial
magnetic field will be switched on along magnetization axis to kick the atom
out of the paramagnetic state. After that it will be switched off. This initial
magnetic field is simply given by the splitting of the potential along
quantization axis, for z-axis, this is simply the intial difference between
spin up and spin down potentials. Note that if the atom already exists in the
magnetic state, this initial splitting will be set to zero automatically unless
this option is suppressed.
To apply external magnetic field along certain direction, specify the value of the field and the axis along which the external field will be switched on. The magnetic field is given in Ry and has a simple interpretation of holding spin-up and spin-down potentials to some constant value if the axis is 0,0,1
D.3 Setting Valence State
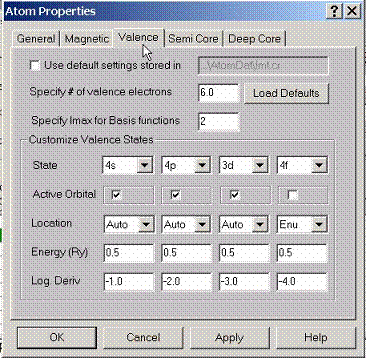 By
default AtomDat directory contains the description of the valence states which
will form the band structure of a given compound. If customization of these settings is
required, this property page can be used.
By
default AtomDat directory contains the description of the valence states which
will form the band structure of a given compound. If customization of these settings is
required, this property page can be used.
D.4 Setting Semicore State
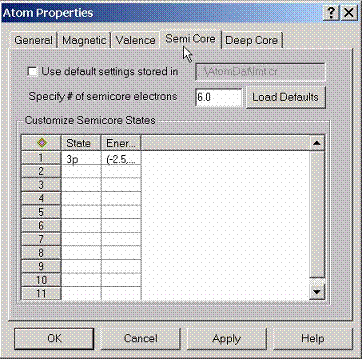 By
default AtomDat directory contains the description of the semicore states which
will form a set of narrow bands below the valence states for a given
compound. If customization of these
settings is required, this property page can be used.
By
default AtomDat directory contains the description of the semicore states which
will form a set of narrow bands below the valence states for a given
compound. If customization of these
settings is required, this property page can be used.
D.5 Setting Deep Core State
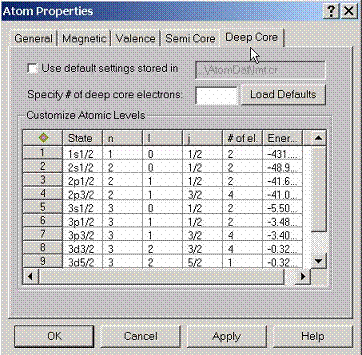 By
default AtomDat directory contains the description of the deep core states
which will be adjusted for a given environment.
If customization of these settings is required, this property page can
be used.
By
default AtomDat directory contains the description of the deep core states
which will be adjusted for a given environment.
If customization of these settings is required, this property page can
be used.
E.1 Setting General Properties for Tight-Binding State
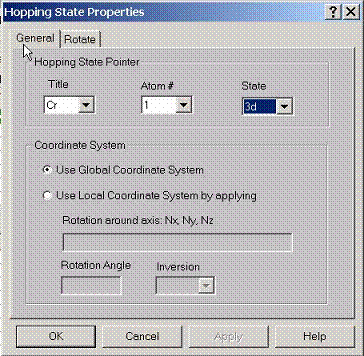 To
calculate hopping integrals between some states, specify pointers to these
states using this property page. Also specify which coordinate system to use
for calculating hopping integrals, either global coordinates or local
coordinates. Local coordinate system, for given atom can be set by rotating
global coordinate system along a certain axis to a certain angle. Also
inversion operation can be applied after such transformation.
To
calculate hopping integrals between some states, specify pointers to these
states using this property page. Also specify which coordinate system to use
for calculating hopping integrals, either global coordinates or local
coordinates. Local coordinate system, for given atom can be set by rotating
global coordinate system along a certain axis to a certain angle. Also
inversion operation can be applied after such transformation.
E.2 Setting Rotate Properties for Tight-Binding State
 If
hopping integrals are known for some local (global) coordinate systems for each hopping state, they can also be
viewed at any other coordinate system. To rotate hopping integrals for one
coordinate system to another coordinate system specify the desired coordinate
system using this property page. Press rotate button which should rotate
the integrals within the hoppings data table to the new coordinate system and
set this coordinate system as the main coordinate system.
If
hopping integrals are known for some local (global) coordinate systems for each hopping state, they can also be
viewed at any other coordinate system. To rotate hopping integrals for one
coordinate system to another coordinate system specify the desired coordinate
system using this property page. Press rotate button which should rotate
the integrals within the hoppings data table to the new coordinate system and
set this coordinate system as the main coordinate system.
E.3 Setting General Properties for Correlated State
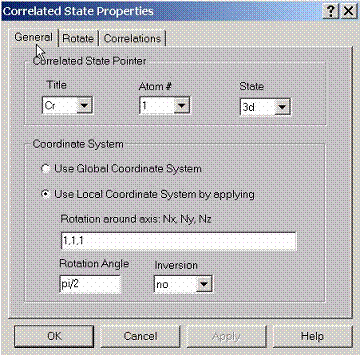 To
treat correlation effects for some states, specify pointers to these states
using this property page. Also specify which coordinate system to use for
calculating density matrices, either global coordinates or local coordinates.
Local coordinate system, for given atom can be set by rotating global
coordinate system along a certain axis to a certain angle. Also inversion
operation can be applied after such transformation.
To
treat correlation effects for some states, specify pointers to these states
using this property page. Also specify which coordinate system to use for
calculating density matrices, either global coordinates or local coordinates.
Local coordinate system, for given atom can be set by rotating global
coordinate system along a certain axis to a certain angle. Also inversion
operation can be applied after such transformation.
E.4 Setting Rotate Properties for Correlated State
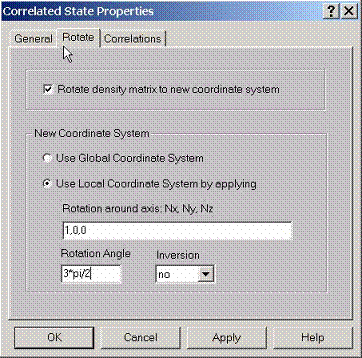 If
density matrices are known for some local (global) coordinate systems for each correlated state, they can also be
viewed at any other coordinate system. To rotate density matrices from one
coordinate system to another coordinate system specify the desired coordinate
system using this property page. Press rotate button which should rotate
the all matrices to the new coordinate system and set this coordinate system as
the main coordinate system.
If
density matrices are known for some local (global) coordinate systems for each correlated state, they can also be
viewed at any other coordinate system. To rotate density matrices from one
coordinate system to another coordinate system specify the desired coordinate
system using this property page. Press rotate button which should rotate
the all matrices to the new coordinate system and set this coordinate system as
the main coordinate system.
E.5 Setting Correlation Properties for Correlated State
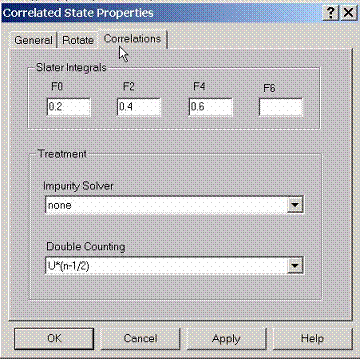 On
site correlation effects have to be described by the Slater integrals. For s
state this is Hubbard constant U or Slater integral F0 which needs to be set.
For p,d, f states there are additional Slater integrals which need to be set.
Since correlations effects are treated on top of the LDA calculation, the
double counting effects have to be taken into account. The most common choice
here is U(n-1/2).
On
site correlation effects have to be described by the Slater integrals. For s
state this is Hubbard constant U or Slater integral F0 which needs to be set.
For p,d, f states there are additional Slater integrals which need to be set.
Since correlations effects are treated on top of the LDA calculation, the
double counting effects have to be taken into account. The most common choice
here is U(n-1/2).
F.1 Accessing Method Controls
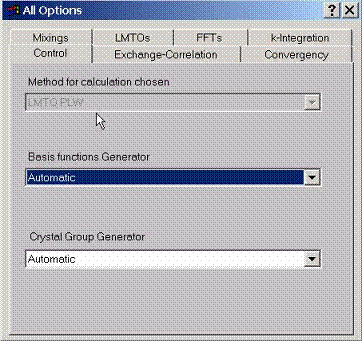 Various
choices to adjust radial basis functions to the atomic potential can be
selected here, they can either be adjusted to (spin dependent) spherical
self-consistent potential, spin-averaged spherical self-consistent potential or
simply found using the input spherical potential and be frozen during the
self-consistent iterations.
Various
choices to adjust radial basis functions to the atomic potential can be
selected here, they can either be adjusted to (spin dependent) spherical
self-consistent potential, spin-averaged spherical self-consistent potential or
simply found using the input spherical potential and be frozen during the
self-consistent iterations.
The BandLab calculates crystal group automatically. In some cases however, one can suppress this option and use only unitary element of the group to perform the calculation. While the advantage of knowledge of the symmetry will not be utilized, this may result in suppressing numerical errors which always appear in comparing the configurations with different symmetries.
F.2 Accessing Method Exchange Correlation Functional
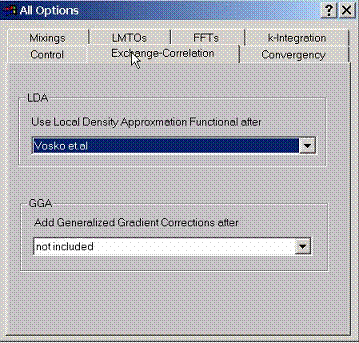 Different
forms of both LDA and GGA exchange correlation functionals can be selected
here.
Different
forms of both LDA and GGA exchange correlation functionals can be selected
here.
F.3 Accessing Method Convergence Criteria
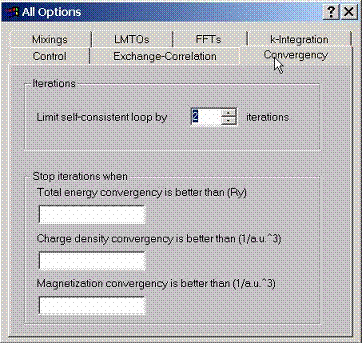 To
limit the total number of iterations, use or interrupt the execution of
self-consistent cycle when one or several criteria are achieved, specify them
using this property page.
To
limit the total number of iterations, use or interrupt the execution of
self-consistent cycle when one or several criteria are achieved, specify them
using this property page.
F.4 Accessing Method Mixing Options
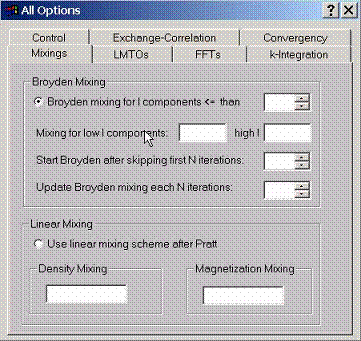 Choose
between linear and Broyden mixing schemes for calculating self-consistent charge densities. Broyden
mixing is usually much faster method to find self-consistent solution but
sometimes maybe unstable. Linear method is the most stable method especially
when the mixing parameters are small but maybe time consuming.
Choose
between linear and Broyden mixing schemes for calculating self-consistent charge densities. Broyden
mixing is usually much faster method to find self-consistent solution but
sometimes maybe unstable. Linear method is the most stable method especially
when the mixing parameters are small but maybe time consuming.
F.5 Accessing LMTO Options
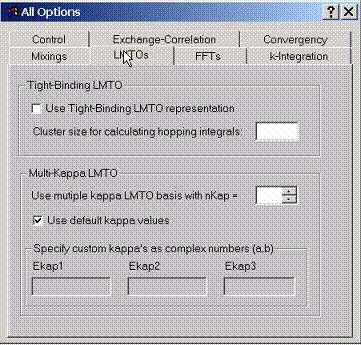 Use
tight binding LMTO representation when calculating the hopping integrals
between various states in order to build the tight-binding model for a given
compound. In this case specify the cluster size within which the hopping
integrals will be calculated.
Use
tight binding LMTO representation when calculating the hopping integrals
between various states in order to build the tight-binding model for a given
compound. In this case specify the cluster size within which the hopping
integrals will be calculated.
Multikappa LMTO basis sets provide additional flexibility to get most accurate solutions of the problem. Usually, when using the Atomic Sphere Approximation, the number of kappa’s (LMTO tails) is set to 1 (default). For full potential LMTO method, the optimal number of kappa’s is 2 (default). Sometimes 3 can be used for most accurate calculations.
Custom tail energies (kappa’s squared) have be specified as complex numbers. For 1, 2, 3 kappa basis sets the default numbers are (-0.1,0.0), (-1.0,0.0), (2.5,0.0). If positive kappa’s are necessary, add some small imaginary part (something like 0.03) to avoid free electron singularities in the structure constants.
F.6 Accessing k Integration Options
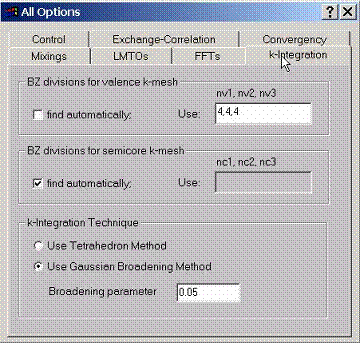 To
perform integrals over k space, select the number of divisions along reciprocal
lattice vectors which will set up the grid. Different grids can be used for
valence and semicore states.
To
perform integrals over k space, select the number of divisions along reciprocal
lattice vectors which will set up the grid. Different grids can be used for
valence and semicore states.
The integration technique can be either tetrahedron method or Gaussian broadening method.
F.7 Accessing Plane wave FFT options
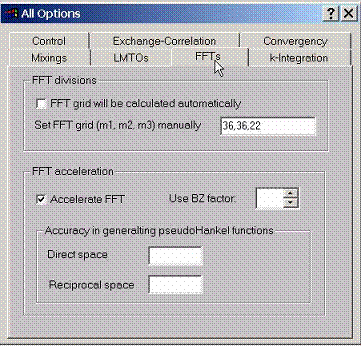 For
a full potential LMTO method, the fast Fourier transform grid can be set
manually. By default the program finds the FFT grid using the criterion of
getting the most accurate solution. It may be too costly however for quick
calculations.
For
a full potential LMTO method, the fast Fourier transform grid can be set
manually. By default the program finds the FFT grid using the criterion of
getting the most accurate solution. It may be too costly however for quick
calculations.
Accelerations of fast Fourier transforms can be achieved for the cost of accuracy.
Accuracy in generating the pseudoHankel functions can be varied (the default value here is 0.02 for direct space and 0.04 for reciprocal space) The smaller this number, the more accurate the calculation for the cost of increasing of the amount of plane waves which will be used to expand pseudoHankel functions in Fourier series.